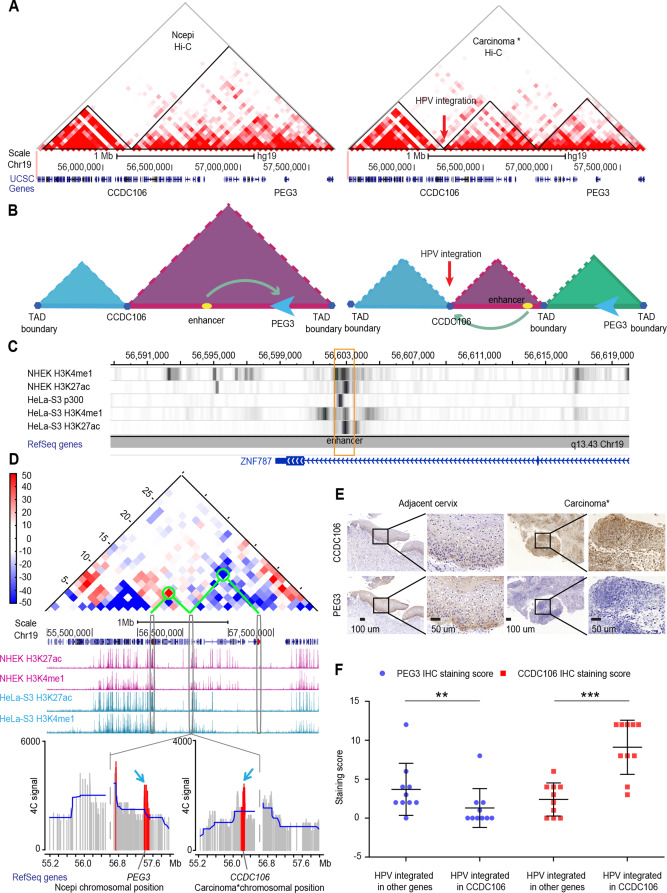
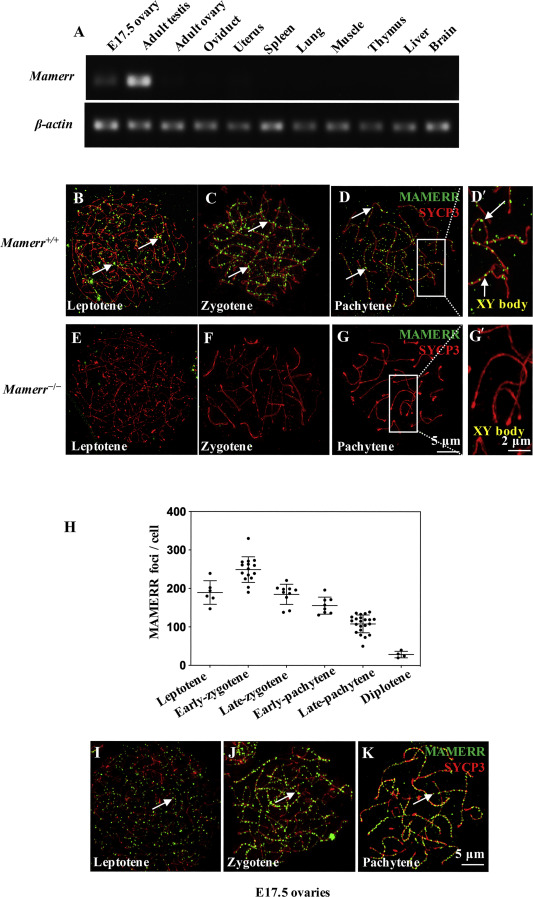
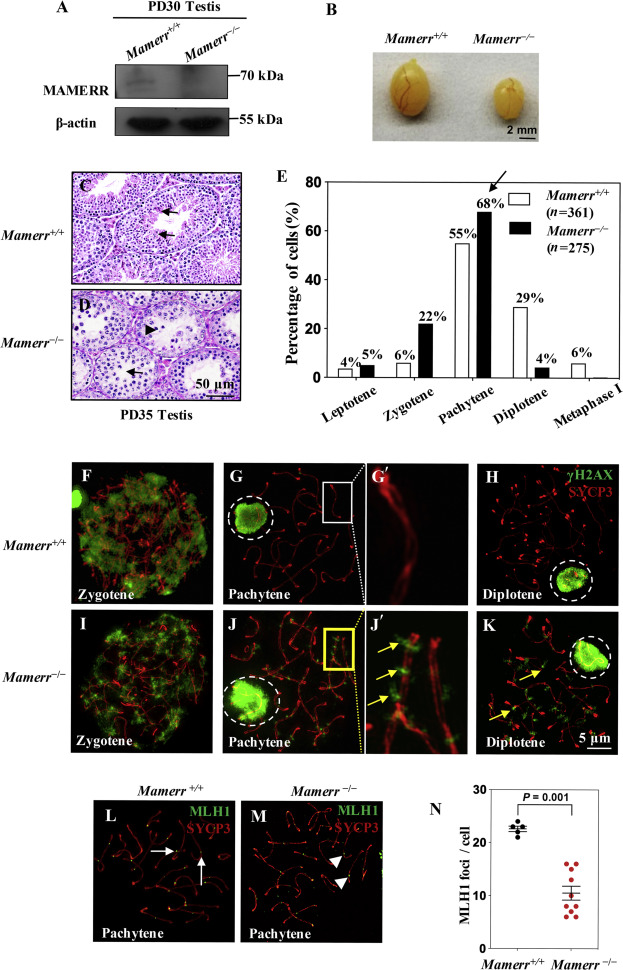
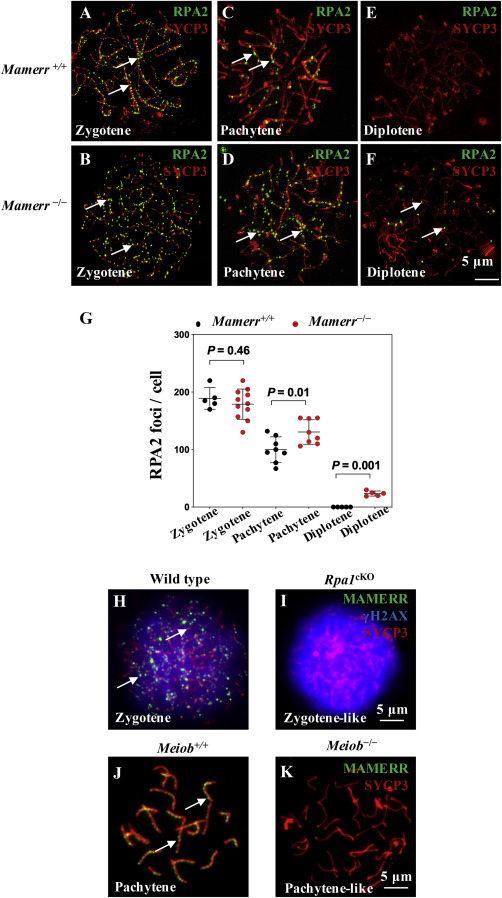
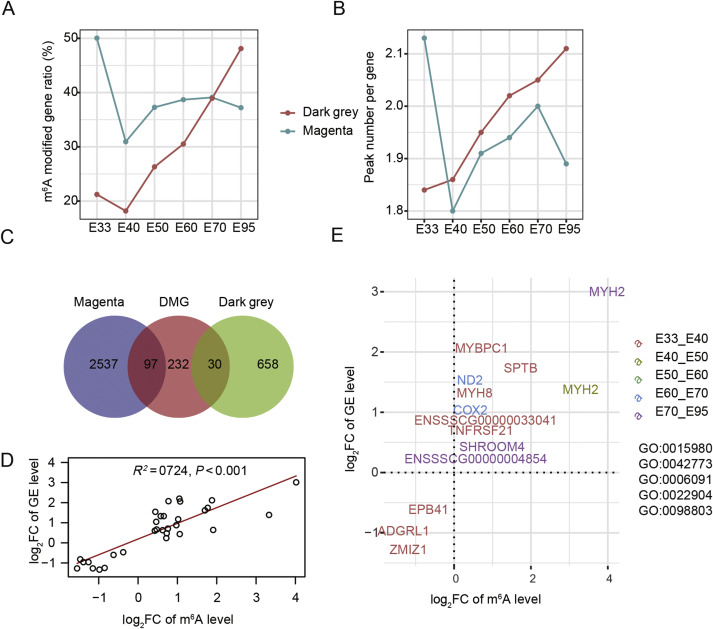
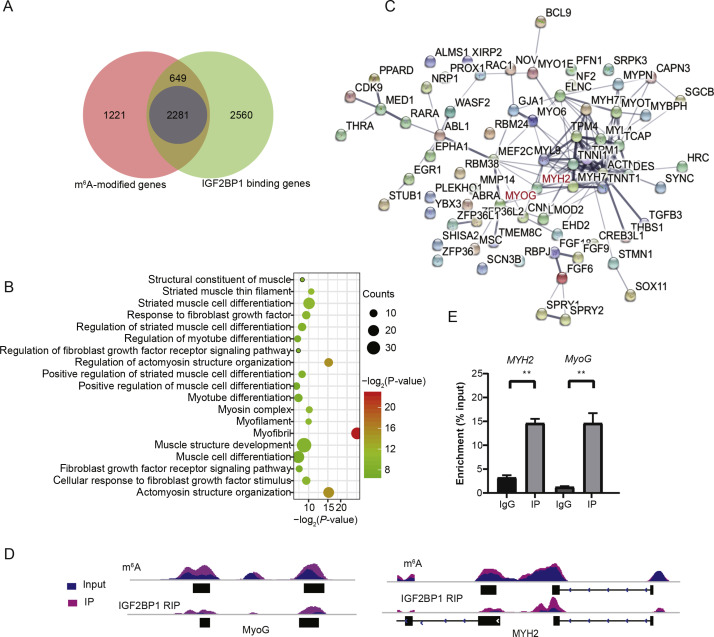
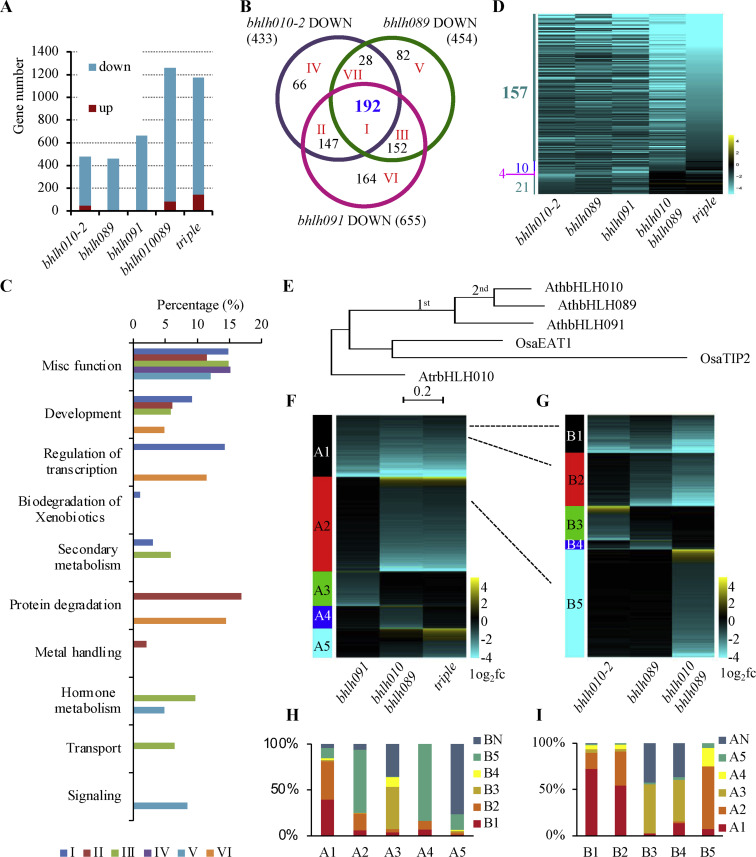
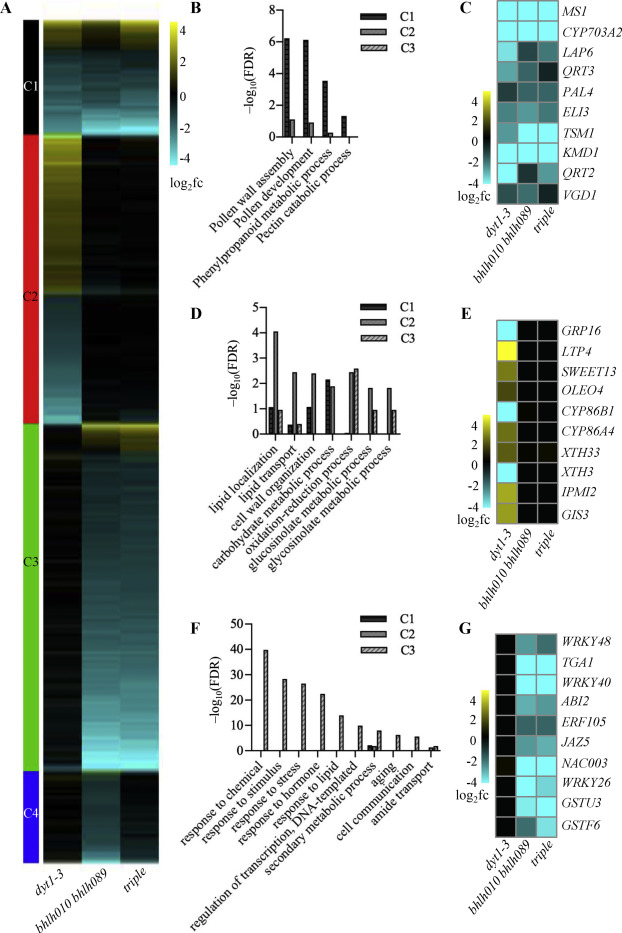
2020, 47(8): 477-492.
doi: 10.1016/j.jgg.2020.09.001
Abstract:
The Arabidopsis bHLH010/089/091 (basic helix-loop-helix) genes are functionally redundant and are required for both anther development and normal expression of DYT1-activated anther-related genes. These three genes are conserved in Brassicaceae, suggesting that each of them is under selection pressure; however, little is known about the possible functional differences among these bHLH genes and between the bHLH and DYT1 genes. Here, we compared novel anther transcriptomic data sets from bHLH010/089/091 single and double mutants, with an anther transcriptomic data set from the wild type (WT) and a previously obtained anther transcriptomic data set from the bhlh010 bhlh089 bhlh091 triple mutant. The results revealed molecular phenotypes that support the functional redundancy and divergence of bHLH010, bHLH089, and bHLH091, as well as the functional overlap and difference between them and DYT1. DNA-binding analyses revealed that DYT1 and bHLH089 specifically recognize the TCATGTGC box to activate the expression of target genes, including ATA20, EXL4, and MEE48. In addition, among genes whose expression was affected in the bhlh010 bhlh089 double and bhlh010 bhlh089 bhlh091 triple mutants, genes that are involved in the stress response and cell signaling were enriched, which included 256 genes whose expression was preferentially induced by heat during early flower development. Moreover, the bhlh double mutants exhibited defective pollen development when the plants were grown under elevated temperature, suggesting that bHLH genes are important for anther gene expression under such conditions. These results are consistent with the observation that the heat-induced expression of several genes is less in thebhlh mutants than that in the WT. Therefore, our results provide important insights into the molecular mechanism underlying the activation of direct targets by DYT1-bHLH089 heterodimers and demonstrate the protective roles of bHLH010/089/091 in maintaining fertility upon heat stress.